![]() Back to Gilead Har’El’s Memorial
Site
Back to Gilead Har’El’s Memorial
Site
From the Servizio di Cardiologia, Ospedale Sant’Andrea, La Spezia, Italy (P.S.); the Howard Hughes Medical Institute and the Cardiovascular Division, Brigham and Women’s Hospital, Boston (C.E.S.); the Department of Cardiological Sciences, St. George’s Hospital Medical School, London (W.J.M.); and the Minneapolis Heart Institute Foundation, Minneapolis (B.J.M.). Address reprint requests to Dr. Spirito at the Servizio di Cardiologia, Ospedale Sant’Andrea, Via Veneto 197, La Spezia 19100, Italy.
Copyright © 1997 by the Massachusetts Medical Society
Recent observations suggest that the prevalence of hypertrophic
cardiomyopathy in the general population is higher (about 1 in 500) than
previously thought. (Ref. 23) The condition therefore appears to be a
common genetic malformation of the heart. The clinical course varies
markedly; some patients remain asymptomatic throughout life, some have
severe symptoms of heart failure, and others die suddenly, often in the
absence of previous symptoms. (Ref. 1-8) The heterogeneous natural history
of the disease and the fact that patients with severe symptoms are
preferentially referred to tertiary care centers have been major
impediments to assembling large study populations that truly represent the
overall spectrum of the disease. Indeed, because most of the literature on
hypertrophic cardiomyopathy is derived from investigations performed at
tertiary care centers, (Ref. 6) the clinical picture of the disease that
has emerged from published studies is profoundly influenced by referral
bias. (Ref. 6, 24-29) This is evident in the fact that the annual mortality
figures for hypertrophic cardiomyopathy from such institutions (3 to 4
percent overall and up to 6 percent in children) (Ref. 1, 3, 5, 30-32) are
substantially higher than those recently reported in unselected populations
(1 percent or less). (Ref. 6, 24-29) These observations suggest that in a
substantial proportion of patients the disease has a more favorable
clinical course than previously believed. Strategies for treatment should
therefore rely on data from relatively unselected populations, as well as
from studies at tertiary care referral centers, and on the clinical
experience of physicians who have focused their investigative efforts on
this disease and acquired particular expertise in its management.
Molecular studies of the genetic alterations responsible for hypertrophic cardiomyopathy also provide insight into the heterogeneity of its clinical features. The disease can be caused by a mutation in one of four genes that encode proteins of the cardiac sarcomere: the β-myosin heavy-chain, cardiac troponin T, α-tropomyosin, and myosin-binding protein C genes (Fig. 1). (Ref. 19-22, 34-37) In addition, mutations in the two genes encoding the myosin light chains have been reported in what appears to be a rare form of hypertrophic cardiomyopathy, (Ref. 38) and other genes that cause the disease are likely to be found. (Ref. 39) This etiologic complexity is further compounded by intragenic heterogeneity; more than 50 disease-causing mutations have been identified in these genes of the sarcomere. (Ref. 19-22, 34-37) Hence, the precise molecular defect responsible for hypertrophic cardiomyopathy often differs in unrelated patients.
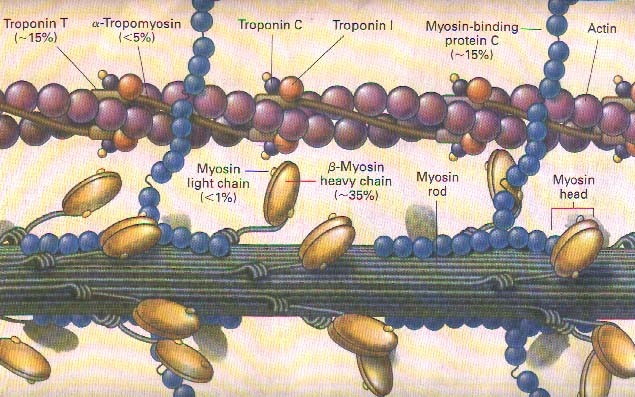
Figure 1. Components of the Sarcomere. Cardiac contraction occurs when calcium binds the troponin complex (subunits C, I, and T) and α-tropomyosin, making possible the myosin-actin interaction. Actin stimulates ATPase activity in the globular myosin head and results in the production of force along actin filaments. Cardiac myosin-binding protein C, arrayed transversely along the sarcomere, binds myosin and, when phosphorylated, modulates contraction. In hypertrophic cardiomyopathy, mutations may impair these and other protein interactions, result in ineffectual contraction of the sarcomere, and produce hypertrophy and disarray of myocytes. Percentages represent the estimated frequency with which a mutation on the corresponding gene causes hypertrophic cardiomyopathy. Modified from Seidman and Seidman. (Ref. 33).
The diverse clinical and genetic features of hypertrophic cardiomyopathy make it impossible to define precise guidelines for management. As in many diseases, it is often necessary to individualize therapy. In hypertrophic cardiomyopathy, the treatment of symptoms to improve quality of life and the identification of patients who are at high risk for sudden death and require aggressive therapy are two distinct issues that must be addressed by largely independent strategies.
Pharmacologic therapy to improve diastolic filling and possibly reduce myocardial ischemia is the primary means of relieving symptoms in hypertrophic cardiomyopathy; it is also the sole therapeutic option for patients without obstruction of the left ventricular outflow, who constitute the great majority of patients with this disease (Fig. 2). (Ref. 3, 5, 7, 8, 40-51) Invasive interventions to abolish the outflow gradient should be considered only for the minority of patients (about 5 percent) who have both marked outflow obstruction and severe symptoms unresponsive to medical therapy. (Ref. 3, 5, 7, 8)
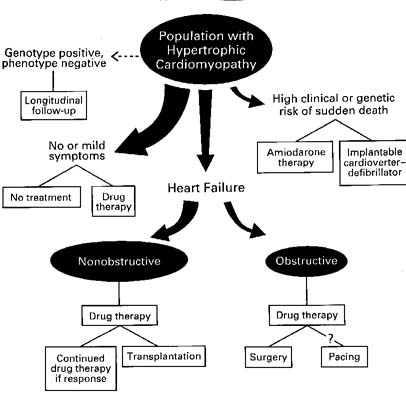
Figure 2. The Principal Clinical Presentations of Hypertrophic Cardiomyopathy and Corresponding Treatment Strategies. The size of the arrows indicates the approximate proportion of patients with hypertrophic cardiomyopathy in each subgroup. The dashed arrow indicates the present uncertainties regarding the size of this subgroup, and the question mark indicates the uncertainties regarding the therapeutic efficacy of pacing. The drugs are specified in the text.
Beta-adrenergic-blocking drugs and verapamil have been used
extensively in the treatment of hypertrophic cardiomyopathy. The beneficial
effects of beta-blockers on symptoms (principally dyspnea and chest pain)
and exercise tolerance appear to be due largely to a decrease in the heart
rate with a consequent prolongation of diastole and increased passive
ventricular filling. (Ref. 45, 52, 53) By reducing the inotropic response,
beta-blockers may also lessen myocardial oxygen demand and decrease the
outflow gradient during exercise, when sympathetic tone is increased. (Ref.
3, 52-54) Verapamil has favorable effects on symptoms because it improves
ventricular filling and probably reduces myocardial ischemia. (Ref. 55-61)
Nifedipine and diltiazem have also been used occasionally in the treatment
of hypertrophic cardiomyopathy. (Ref. 62-64) Because of its potent
vasodilative effects, however, nifedipine may be harmful, particularly in
patients with outflow obstruction. (Ref. 65)
Disopyramide may reduce the gradient and relieve symptoms by virtue of its negative inotropic properties (Ref. 66); in many patients, however, the initial hemodynamic and clinical benefits decrease with time. (Ref. 3, 8) Because disopyramide may shorten the atrioventricular nodal conduction time and thus increase the ventricular rate during paroxysmal atrial fibrillation, supplementary therapy with beta-blockers in low doses is advisable.
In patients with hypertrophic cardiomyopathy, beta-blockers and
verapamil have traditionally been administered on an empirical basis,
relying on the patient’s subjective perception of benefit. Such judgments,
however, are often difficult to make given the day-to-day variability of
patients’ symptoms. Treadmill exercise testing and measurements of maximal
oxygen consumption have proved helpful in determining which patients may
benefit from pharmacologic therapy. (Ref. 67)
Drug selection is not standardized and is based, in part, on the
experience and preferences of individual investigators. Most favor
beta-blockers over verapamil as the initial treatment for exertional
dyspnea, although it is not of critical importance which drug is used
first. Some investigators confine the administration of verapamil to
patients without outflow obstruction and use beta-blockers, disopyramide,
or both for patients with obstruction; others administer verapamil if chest
pain is the predominant symptom. There is no evidence that the
administration of beta-blockers and verapamil together is more advantageous
than the use of either drug alone.
Patients who have no response to beta-blockers often have symptomatic improvement if given verapamil. (Ref. 5, 8, 55, 68) In some patients, this may be due in part to the withdrawal of the beta-blockers and the abolition of their associated side effects. (Ref. 69) In patients with a substantial outflow gradient or markedly elevated pulmonary pressure (or both), verapamil should be used with caution, because the drug’s vasodilative effects may lead to serious hemodynamic complications. (Ref. 5, 8, 70) The symptoms of patients who have congestive heart failure despite treatment with beta-blockers or verapamil may improve with the addition of a diuretic agent. (Ref. 3, 5, 8, 71) However, because many of these patients have diastolic dysfunction and require relatively high filling pressures to achieve adequate ventricular filling, it is advisable to administer diuretics with caution.
In recent years, molecular genetic studies have enabled physicians to
identify an increasing number of asymptomatic patients. (Ref. 19-22, 34-37)
In addition, studies of relatively unselected populations not followed at
referral centers have shown that many patients with hypertrophic
cardiomyopathy have mild or no symptoms. (Ref. 6, 24-29) Taken together,
these observations suggest that relatively asymptomatic patients probably
constitute the majority of the overall patient population (Fig. 2).
There is no evidence that either beta-blockers or verapamil protects
patients with hypertrophic cardiomyopathy from sudden death. (Ref. 3, 5, 8)
Whether these drugs should be used prophylactically to delay disease
progression and improve the prognosis in asymptomatic patients has also
been a subject of debate for many years. The effectiveness of prophylactic
treatment has not been tested prospectively because study populations are
small and the traditional end points (clinical deterioration and premature
death) are infrequent. A further element of uncertainty is the growing
awareness that an important proportion of patients with hypertrophic
cardiomyopathy remain asymptomatic for many years and often have normal
longevity. (Ref. 6, 24-29, 72-74) Therefore, treatment aimed at preventing
the progression of disease does not appear justified in most asymptomatic
patients. One possible exception may be patients with massive left
ventricular hypertrophy (a maximal wall thickness of 35 mm or more in
adults, or a comparable value in children). (Ref. 75, 76) The presence of
such striking morphologic features usually represents a strong motivation
to initiate pharmacologic treatment, even in the absence of symptoms. This
decision is probably appropriate and is justified by the expectation that
in the presence of massive hypertrophy the favorable effects of drugs on
diastolic filling and myocardial ischemia might delay the onset of
symptoms.
Asymptomatic children or young adults with particularly marked outflow tract gradients constitute another subgroup in which physicians often feel the need to initiate prophylactic drug treatment. Although systematic data are not available on the effects of prophylactic therapy in this subgroup, it may be appropriate to initiate drug treatment with the expectation that it might reduce the hemodynamic burden and improve the clinical course of the disease.
Atrial fibrillation is a particularly important arrhythmia in hypertrophic cardiomyopathy because it develops in a substantial proportion of adult patients (Ref. 28, 77) and is associated with an increased risk of systemic thromboembolism, heart failure, and death (Fig. 3). (Ref. 3, 5, 8, 77-80) Paroxysmal episodes of atrial fibrillation often cause rapid clinical deterioration by reducing diastolic filling and cardiac output, usually as a consequence of the high ventricular rate. Conversely, chronic atrial fibrillation is often well tolerated if the heart rate is adequately controlled.
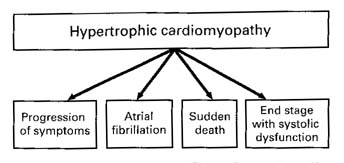
Figure 3. The Principal Pathways of Disease Progression in Hypertrophic Cardiomyopathy.
Amiodarone is probably the most effective antiarrhythmic agent for the prevention of recurrences of atrial fibrillation. (Ref. 77, 78, 81-83) Beta-blockers and verapamil are usually efficacious in controlling the heart rate in patients with chronic atrial fibrillation, (Ref. 3, 5, 8, 77) but ablation of the atrioventricular node and the implantation of a pacemaker may be necessary in selected patients. Anticoagulant therapy is indicated in patients with recurrent or chronic atrial fibrillation. Since recurrences of even brief episodes of atrial fibrillation in hypertrophic cardiomyopathy have been associated with a substantial risk of systemic embolization, (Ref. 77, 79) the threshold for the initiation of anticoagulant therapy should be low.
In some patients, medical therapy ultimately proves insufficient to
control symptoms. At this point in the clinical course of the disease,
subsequent therapeutic strategies are largely determined by the presence or
absence of outflow obstruction (Fig. 2).
Patients without outflow obstruction and with severe heart failure have more limited therapeutic options. An important minority of these patients have arrived at an end stage characterized by left ventricular remodeling with progressive wall thinning (due to myocardial necrosis), cavity enlargement, and systolic impairment (Fig. 3). (Ref. 84, 85) In such patients, the treatment should be changed from beta-blockers or verapamil to the standard therapeutic agents for heart failure associated with systolic dysfunction, including diuretics, angiotensin-converting-enzyme inhibitors, and digitalis. Ultimately, these patients may become candidates for heart transplantation and are probably the only subgroup, within the broad spectrum of the disease, in which transplantation should be considered. (Ref. 86)
The small subgroup of patients who have both a large outflow gradient
(50 mm Hg or more) and severe symptoms of heart failure that are
unresponsive to medical treatment are candidates for surgery. (Ref. 3, 5, 8)
Whether patients with a marked gradient only under provokable conditions
should also be considered candidates for surgery is unclear.
Usually, surgical reduction of the outflow gradient is achieved by
removing a small amount of muscle from the basal septum (myotomy-myectomy).
(Ref. 87-96) Replacement of the mitral valve with a low-profile prosthesis
has also been used as an alternative to myotomy-myectomy (Ref. 90, 97, 98) in
selected patients. These have included patients with severe mitral
regurgitation due to intrinsic abnormalities of the valve apparatus, such
as prolapse; patients with mid-cavity obstruction due to anomalous
insertion of papillary muscle into the anterior mitral leaflet (Ref. 99);
and patients with only mild septal hypertrophy, which suggests that
muscular resection would be associated with a high risk of septal
perforation or an inadequate hemodynamic result. (Ref. 90, 98) Mitral
valvuloplasty has been combined with myotomy-myectomy in some patients with
particularly elongated mitral leaflets. (Ref. 100-102)
Surgery abolishes or substantially reduces the basal outflow gradient
in more than 90 percent of patients. (Ref. 87-96) Published data on about
1500 patients from North American and European centers reveal substantial
and persistent symptomatic improvement in about 70 percent of patients five
or more years after operation. (Ref. 88-96) Because myotomy-myectomy
requires extensive surgical experience, operative mortality is lowest (less
than 2 percent) at the centers where the most operations are performed.
(Ref. 8, 89, 91, 93, 95, 96) Surgical mortality appears to be higher among
elderly patients and those undergoing both myotomy-myectomy and
coronary-artery bypass grafting. (Ref. 96, 103, 104) Intraoperative
echocardiography is helpful in determining the site and extent of the
myotomy-myectomy and defining the mitral-valve structure. (Ref. 105, 106)
The mechanisms by which operation improves cardiac function and
relieves symptoms are poorly understood, but they probably arise from the
interaction of several pathophysiologic variables. (Ref. 107-113) A marked
reduction or normalization of the intraventricular systolic and
end-diastolic pressures is the most tangible consequence of surgery, which
may also have a favorable influence on left ventricular filling and
myocardial ischemia. (Ref. 3, 5, 8, 107, 112, 113)
Surgery is not performed in asymptomatic or mildly symptomatic patients for a number of reasons: the effect of surgery on survival is unknown (Ref. 88-96); outflow obstruction can be compatible with normal longevity (Ref. 6, 24-29, 72-74); and in some patients obstruction is not the sole functional abnormality, and surgery therefore cannot be regarded as curative. (Ref. 1-3, 5, 8)
In recent years, there has been growing interest in the potential use
of dual-chamber pacing as an alternative to surgery in patients with
hypertrophic cardiomyopathy. (Ref. 14-18) Some studies have reported that
dual-chamber pacing is associated with both a substantial decrease in the
outflow gradient and symptomatic improvement in patients with outflow
obstruction and severe functional limitation who are unresponsive to drug
treatment. (Ref. 16, 17) These observations, however, come from
nonrandomized, unblinded studies and are based on the subjective perception
of symptomatic improvement reported by patients over a relatively short
period.
Recent and more carefully controlled investigations have found the
effects of dual-chamber pacing in hypertrophic cardiomyopathy to be less
favorable. In a randomized, double-blind, crossover study, the average
decrease in the outflow gradient during pacing was small (only about 25
percent) and varied substantially among patients. (Ref. 114) Subjective
symptomatic improvement was reported with similar frequency by patients
after two to three months of pacing and after the same period without
pacing. Objective measurements of exercise capacity (e.g., maximal oxygen
consumption) with and without pacing did not differ significantly. These
findings suggest that a placebo effect may play an important part in the
short-term symptomatic improvement reported by patients. Moreover, two
other studies have shown that a decrease in the outflow gradient produced
by temporary atrioventricular sequential pacing may be associated with
detrimental effects on ventricular filling and cardiac output. (Ref.
115, 116)
The mechanisms by which pacing reduces the outflow gradient are not
understood. Preexcitation of the right ventricle would appear to have a
role, by altering the synergy of ventricular contraction. (Ref. 14-18)
Thinning of the left ventricular wall after pacing was reported in one
study, (Ref. 17) but the observation has been a subject of controversy and
requires independent validation. (Ref. 117) It should also be underscored
that pacing is substantially more complex in hypertrophic cardiomyopathy
than in other cardiac diseases; to produce and subsequently maintain a
decrease in the gradient requires the achievement of maximal right
ventricular preexcitation, both at rest and during exercise, without
compromising ventricular filling and cardiac output. Usually, the
prolongation of atrioventricular conduction time by beta-blockers or
verapamil — or ablation of the atrioventricular node in selected instances
-- is required to achieve this purpose. (Ref. 17, 18)
The role of dual-chamber pacing for the relief of symptoms in
obstructive hypertrophic cardiomyopathy is unclear, and a measured approach
should be adopted in the use of this therapy. In addition, there is no
evidence that pacing reduces the risk of sudden death or alters the
clinical course of the disease.
Recently, the injection of alcohol into the first major septal coronary artery has been proposed as a means of reducing both septal thickness and the outflow gradient in patients with hypertrophic cardiomyopathy. (Ref. 118) Experience with this technique, however, is as yet very limited.
Asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy may die suddenly and unexpectedly. (Ref. 1-3, 5, 7, 8, 119, 120) Stratification of risk has been focused on younger patients, since the achievement of advanced age itself testifies to a more benign form of the disease, for which aggressive management is usually not justified. Despite intense investigation, however, identification of the individual patient at high risk remains a major challenge. This is due, in part, to the fact that most data on risk stratification have been assembled at referral institutions and are based on patient populations already highly selected in terms of risk. (Ref. 6) The multiplicity of mechanisms believed to cause sudden death in hypertrophic cardiomyopathy and the uncertainties regarding their relative importance constitute other obstacles to risk stratification. The available data suggest that ventricular tachyarrhythmias are the cause of sudden death in most patients, either as a primary event related to the arrhythmogenic myocardial substrate or as a secondary phenomenon triggered by myocardial ischemia, diastolic dysfunction, outflow obstruction, systemic arterial hypotension, or supraventricular tachyarrhythmias. (Ref. 3, 5, 7, 8, 32, 81, 120, 126) In particular, it has been speculated that life-threatening arrhythmias could be triggered by a vicious circle of increasing ischemia and diastolic and systolic dysfunction leading to a decrease in stroke volume and coronary perfusion. Bradyarrhythmias due to sinus-node dysfunction or atrioventricular block may also be responsible for sudden death in some patients. (Ref. 127-129)
No single test can reliably predict risk in hypertrophic
cardiomyopathy, and the complexity of the disease continues to be an
important obstacle to risk stratification. Despite the difficulties in the
assessment of prognosis, most investigators agree that some patients are at
particularly high risk for sudden death and should be treated aggressively
(Fig. 2) (Ref. 3, 5, 7, 8, 19, 22, 32, 120, 130): the rare patients who survive a
cardiac arrest with documented ventricular fibrillation or who have
episodes of sustained ventricular tachycardia, and young patients with a
substantial family history of sudden death (defined as a cluster of two or
more sudden deaths in young family members) or a high-risk mutation.
When such patients are removed from the overall patient population, it
becomes more difficult to identify other subgroups that can be considered
at sufficiently high risk to justify prophylactic treatment. Of particular
relevance is the issue of nonsustained ventricular tachycardia. The
identification of infrequent bursts, or even a single short burst, of
ventricular tachycardia on 24-to-48-hour ambulatory electrocardiographic
monitoring has been considered a justification for antiarrhythmic
treatment. (Ref. 78, 123) However, most of these patients are not at high
risk, (Ref. 27) and intramyocardial conduction studies have shown that many
do not have electrical instability. (Ref. 131) As a result, the
recommendation that amiodarone or other antiarrhythmic drugs be given to
all patients with nonsustained ventricular tachycardia has been withdrawn.
On the other hand, it is reasonable to assume that frequent or prolonged
episodes of nonsustained ventricular tachycardia on a single Holter
recording (arbitrarily defined here as more than five episodes or a run of
10 or more beats) may suggest a high risk of sudden death and should be
considered a justification for prophylactic treatment. (Ref. 27)
Studies in tertiary referral centers have suggested that several other features may be associated with an increased risk of sudden death, including the onset of symptoms in childhood, particularly marked hypertrophy, and exercise-induced hypotension. (Ref. 32, 119, 125, 126, 132, 133) These variables have a low positive predictive accuracy and cannot reliably identify individual patients for whom an aggressive therapeutic strategy may be warranted. (Ref. 32, 125, 126, 133) Their negative predictive accuracy, however, is relatively high. Therefore, the absence of certain clinical characteristics can be used to develop a profile of patients at low risk for sudden death. Adult patients can be considered at low risk if they have no symptoms or mild symptoms and also have none of the following: a family history of premature death due to hypertrophic cardiomyopathy, nonsustained ventricular tachycardia during Holter monitoring, a marked outflow tract gradient, substantial hypertrophy (more than 20 mm), marked left atrial enlargement, and an abnormal blood-pressure response during exercise. Such low-risk patients constitute an important proportion of the overall population of patients with hypertrophic cardiomyopathy and are frequently encountered at nonreferral centers or during routine screening of affected pedigrees. (Ref. 6, 24-29) These patients do not need medical treatment and should be reassured about their prognosis; they require little or no restriction with regard to recreational sports and employment. However, because hypertrophic cardiomyopathy is the most common cause of sudden death in young athletes, it is recommended that all patients with the disease avoid intense training and competition. (Ref. 134-137)
There is no convincing evidence that electrophysiologic testing has an important role in identifying the patients with hypertrophic cardiomyopathy who are at high risk for sudden death. Standard programmed stimulation with two ventricular premature depolarizations seldom induces sustained ventricular tachycardia even in patients with hypertrophic cardiomyopathy who are known to be at high risk, such as survivors of cardiac arrest. (Ref. 138-140) Conversely, more aggressive protocols, with three ventricular premature depolarizations, frequently trigger polymorphic ventricular tachycardia or ventricular fibrillation even in patients with hypertrophic cardiomyopathy who are at low risk for sudden death (Ref. 139, 140); on the basis of experience in coronary artery disease, dilated cardiomyopathy, and hypertrophic cardiomyopathy, these latter arrhythmias are generally regarded as nonspecific responses. (Ref. 131, 139, 141) The induction of such arrhythmias in a patient thus does not justify aggressive treatment.
The prognostic importance of syncope in hypertrophic cardiomyopathy is
not completely understood, and the diversity of the mechanisms potentially
responsible for this symptom makes management particularly complex. (Ref.
126-129, 142-144) These uncertainties have often led to the assumption that
even a single syncopal episode is equivalent to aborted sudden death and
may justify aggressive therapy. However, it should be emphasized that
isolated and remote episodes of syncope are not in-frequently reported by
patients (Ref. 27, 28) and that a history of recent syncope is uncommon in
patients who have died suddenly. (Ref. 119) Furthermore, it is usually
impossible to determine retrospectively the mechanism responsible for a
single episode of syncope in a given patient. Therefore, patients with a
single syncopal episode should be evaluated conservatively with noninvasive
testing, including Holter monitoring, patient-activated event detectors,
and the assessment of blood-pressure response during exercise.
Patients with recurrent syncope are more problematic; their treatment constitutes one of the most difficult challenges in the management of hypertrophic cardiomyopathy. The treatment of such patients must be individualized and designed according to the potential role of each of the diverse mechanisms that may be responsible for syncope, including ventricular and supraventricular tachyarrhythmias, bradyarrhythmias, marked outflow obstruction, autonomic dysfunction, and complex interactions between myocardial ischemia and diastolic dysfunction that may lead to a sudden and marked decrease in cardiac output.
Massive hypertrophy (defined as a maximal wall thickness of 35 mm or more in adults and the equivalent in children) is part of the morphologic spectrum of hypertrophic cardiomyopathy. (Ref. 75, 76) Such dramatic phenotypic expressions, however, are virtually never encountered in patients more than 50 years old, implying that most patients with these structural abnormalities die prematurely or undergo substantial left ventricular remodeling with wall thinning. (Ref. 84, 85, 145) The risk of sudden death associated with massive hypertrophy is uncertain. Nevertheless, the coexistence of this morphologic characteristic and one or more of the risk factors discussed above should arouse great concern and serve as an indication for prophylactic treatment.
A recent approach to risk stratification in hypertrophic
cardiomyopathy has focused on the impact of genotype on clinical outcome.
The available data suggest that β-myosin heavy-chain mutations may
account for approximately 30 to 40 percent of cases of familial
hypertrophic cardiomyopathy. (Ref. 19-21, 34-37) The prognosis for patients
with different myosin mutations varies considerably. Some myosin mutations
are benign (e.g., Val606Met), whereas others are associated with premature
death from either heart failure or sudden catastrophic events. (Ref. 19-21)
The Arg403Gln mutation appears to be associated with markedly reduced
survival. In families with this mutation, no more than 50 percent of the
affected family members survived past 45 years of age. (Ref. 19) Cardiac
troponin T mutations are estimated to account for about 10 to 20 percent of
cases of familial hypertrophic cardiomyopathy. (Ref. 22, 34) The clinical
manifestations associated with the eight reported cardiac troponin T
mutations are similar. The cardiac hypertrophy is relatively mild and can
be subclinical in some adults, but life expectancy is substantially
reduced. (Ref. 22, 34) Collectively, these observations suggest that genetic
data may have an increasing influence on the management of this disease.
Although at present the identification of disease-causing mutations remains
a complex and time-consuming endeavor, the identification of a genetic
defect that is associated with adverse outcomes should prompt careful
clinical assessment and intervention.
Genetic diagnosis will also lead to the identification of increasing
numbers of persons with preclinical hypertrophic cardiomyopathy (that is,
with a genetic defect but no phenotypic alteration). The clinical
implications of such genetic abnormalities have not been determined, and
there is no evidence that assessment of the risk of sudden death or
pharmacologic treatment is indicated, unless a malignant mutation is
identified.
Although there is intense interest in genetic therapy for inheritable human conditions, the immediate application of this technology to hypertrophic cardiomyopathy is problematic. The disease is transmitted as an autosomal dominant trait, and affected persons have one mutated and one normal allele. Since most mutations cause the substitution of a single amino acid within the encoded protein, appropriate therapy would have to selectively inactivate the mutated gene, the encoded protein, or both.
In patients with hypertrophic cardiomyopathy who are considered to be at particularly high risk for life-threatening tachyarrhythmias, the available therapeutic options for the prevention of sudden death are the same as in coronary artery disease or dilated cardiomyopathy — that is, amiodarone or the implantable cardioverter-defibrillator. (Ref. 9-12, 146, 147) The precise clinical criteria for choosing between these two treatment strategies have not yet been defined. A number of nonclinical factors may influence the choice, such as the availability of advanced technology and cost-benefit considerations. Furthermore, cultural differences in patients’ expectations and physicians’ attitudes may have an important influence on the decision. For example, in the United States, cultural factors and available resources predispose society to an aggressive pursuit of all therapeutic options; in many European countries more conservative strategies prevail.
The use of amiodarone for the prevention of sudden death in hypertrophic cardiomyopathy is based on the results of a nonrandomized study, (Ref. 148) as well as on inferences drawn from other cardiac diseases and concern about the potential proarrhythmic effects of other antiarrhythmic drugs. (Ref. 146-148) Relatively low doses of amiodarone (100 to 300 mg per day) have been used in patients with hypertrophic cardiomyopathy who are judged to be at high risk for life-threatening tachyarrhythmias. (Ref. 81, 149) In patients with advanced disease and severe heart failure, higher doses may be associated with higher mortality. (Ref. 150, 151)
Systematic data are not currently available on the effectiveness of
the implantable cardioverter-defibrillator in hypertrophic cardiomyopathy.
It is a reasonable expectation, however, that the device will be effective
in terminating life-threatening tachyarrhythmias. In addition, the pacing
capabilities of the most recent defibrillators offer protection from
cardiac arrest due to bradyarrhythmias. Therefore, the implantable
defibrillator represents an important option for patients in the high-risk
subgroups. The availability of a device with the potential to abort lethal
arrhythmias and preserve life also presents clinicians with difficult
decisions regarding which other patients should be offered this treatment.
Because of the potential complications associated with the defibrillator,
(Ref. 11, 12) as well as the awareness that the vast majority of patients
will not die suddenly, we resist extending this therapy to patients other
than those who can be definitively judged to be at high risk. Nevertheless,
because of the imprecision of risk stratification, we believe that it would
be unethical to withhold information about either the defibrillator or
amiodarone from patients who are perceived to be at high risk, but in whom
the prognosis cannot be reliably determined.
Because of the persistent uncertainties, the prevention of sudden death in patients with hypertrophic cardiomyopathy continues to be an important challenge. Although many compelling questions remain unanswered, more definitive recommendations will undoubtedly emerge as new data become available.
Supported by grants from the Consiglio Nazionale delle Ricerche and Telethon-Italia (to Dr. Spirito), the National Institutes of Health and Howard Hughes Medical Foundation (to Dr. Seidman), the British Heart Foundation (to Dr. McKenna), and the Minneapolis Heart Institute Foundation (to Dr. Maron).
We are indebted to Dr. Liviu Poliac and Dr. Donald Fischman for assistance in preparing the figures.